A ló származása, és a lófajták genetikai rokonsága
A rokonsági és leszármazási viszonyok megállapítása során olyan DNS szakaszokat hasonlítunk össze melyek funkcióját nem ismerjük, de tudjuk, hogy azok a fajon belül változatosságot mutatnak, egyedenként változóak. Minél több egyednek ismerjük a teljes genom szekvenciáját annál több változékony szekvencia darab azonosítható, és annál pontosabban tisztázhatók a rokonsági viszonyok. A módszer lényege tehát az, hogy összehasonlítjuk nagyszámú, különböző fajtából és földrajzi régióból származó ló teljes DNS szekvenciáját. Technikai és pénzügyi okokból elsőként a mitokondriális DNS szekvenciákat szokás meghatározni, melynek segítségével közvetlenül azonosíthatók a leszármazási vonalak. A lovak mtDNS adatai a 2000-es évek során kezdtek felhalmozódni, és több szempontból is igen meglepő eredményt adtak.
Anyai ágú leszármazási vonalak vizsgálati eredményei:
1., A házilovak nagyszámú anyai vonalba tartoznak, ami arra utal, hogy a többi háziállattal ellentétben igen nagyszámú vadló kanca került háziasításra, valószínűleg a vadló 6000 évvel ezelőtti elterjedési területének különböző pontjain. A mai lovak közel ötven anyai vonalba tartoznak, és a háziasítás kezdete óta kihalt vonalakat figyelembe véve a háziasítás 100 körüli kancát érinthetett (Cieslak 2010, Lippold 2011).
2., Az anyai vonalak elsoszlása nem köthető sem fajtához, sem földrajzi régióhoz. A legkülönbözőbb fajtákban is ugyanazon ősanyák leszármazottait találjuk, és az ázsiai lovakban ugyazok az anyai vonalak vannak jelen mint az európai lovakban (Achilli 2012). Az mtDNS alapján még a hidegvérű és melegvérű vonalak sem különíthetők el, vagy a pónik sem egymástól sem a többi fajtától (Lippold 2011, Bigi 2014) .
Ennek egyértelmű olvasata az, hogy a házilovakra NEM az volt jellemző, hogy az utódok többsége a szülők közelében maradt. Ehelyett a lovak anyai vonalai igen gyorsan terjedtek el kontinensnyi távolságokban ami nyilvánvalóan a ló felhasználásából, és gyorsaságából következik. A kívánatos tulajdonságokat mutató egyedek utódai hamar óriási távolságokra kerültek az eredeti származási helyüktől. Ma közel 1000 lófajtát különböztetünk meg, de nem szabad elfelejteni, hogy a legtöbb fajta tenyésztése az 1700-as években vette kezdetét. DNS vizsgálatok még a nagymúltú arab lovakról is kimutatták, hogy egyetlen tenyészvonal utódai nagyszámú anyaállatra vezethetők vissza, másrészt ugyanazon ősanyák leszármazottai különböző tenyészvonalakban is jelen vannak (Khanshour 2013). Hajdanán valószínűleg csak az egyedek minősége számított, származástól függetlenül.
3., Az adatokból kiderült, hogy egyetlen lófajta sem a Przewalski ló leszármazottja, mert a két csoport anyai vonalai között nincs átfedés. Minden háziló – akár taki, akár tarpán típusú – a tarpán leszármazottja (Jansen 2002).
Apai ágú leszérmazási vonalak vizsgálati eredményei:
Az Y-kromoszóma DNS szekvenciájának vizsgálata technikailag nehezebb és költségesebb mint az mtDNS-é, de nemrég erre is fény derült, és az eddigiek ismeretében még meglepőbb eredményt hozott. Az eddig megvizsgált több mint 100 különböző alapító vonalhoz tartozó hím egyed egyetlen háziasított ősapa leszármazottjának bizonyult (Wallner 2013, Brandariz-Fontes 2013), és csupán a kínai lóállományban sikerült nemrég kimutatni ettől eltérő apai vonalakat (Han 2015). A Przewalski lovak apai vonalai élesen elkülönülnek a házilóétól, ami tovább erősíti, hogy az nem őse a házilónak. Az apai vonalak alacsony száma részben a ló természetes szaporodási módjának tudható be (hárem), amit a tenyésztés méginkább beszűkít a tenyészcsődörök szigorú szelekciójával. Az eredmények arra utalnak, hogy a kancákkal ellentétben a háziasítás igen alacsony számú hím egyedből indulhatott ki, valószínűleg a mének nehezebb szelídíthetősége korlátozta a háziasítható egyedek számát.
Az apai és anyai vonalakból származó információk együttesen arra engednek következtetni, hogy a nyugat-eurázsiai sztyeppéről származó első háziasított lóállomány egyedeit a hadi és gazdasági előnyök miatt Eurázsia szerte hamar importálták, majd a helyi vadló állomány kancáival egészítették ki. A régészeti feltárások arra utalnak, hogy 6000 évvel ezelőtt a vadló Európa nagyrészén ritka lehetett, csak az ibériai félszigeten maradt számottevő állomány. Az mtDNS vizsgálatok ezt megerősítik, mivel a spanyol (és európai) lófajtákban néhány haplocsoport gyakoribb mint az ázsiai vonalakban, ezzel ellentétben a legtöbb haplocsoport ezzel ellenkező gyakoriság eloszlást mutat (Achilli 2012). A háziló kezdeti elterjedését követően a helyi lóállományok a történelem során továbbra is kontinens-szerte intenzíven keveredtek, így egyetlen régióban sem rögzülhettek területre vagy fajtára jellemző genetikai vonalak.
A házilóló származása:
Láttuk, hogy az apai és anyai ágú leszármazási vonalak genetikai vizsgálata nem adott választ arra, hogy hol lehetett a háziasítás kezdete. Régészeti adatokból tudjuk, hogy ez az Urál hegység lábától a mai Észak-Kazahsztánig terjedő területen lehetett, amit a további genetikai vizsgálatok is megerősítettek. A kutatók a többi kromoszómán lévő DNS szakaszok (lásd ábra) legváltozékonyabb, úgynevezett mikroszatellita szekvenciáit kezdték vizsgálni. Azt találták, hogy a lovak örökítőanyaga az egymástól 7000 km-re lévő területeken is meglepően hasonló, ami megerősíti a lóállomány intenzív keveredését. Ezzel a módszerrel azonban egyértelműen kimutatható volt a genetikai diverzitás csökkenése keletről nyugati irányban. Magyarán az ázsiai lovak genetikailag sokfélébbek, míg az európai állomány ettől egységesebb. Ez arra utal, hogy az elterjedés keletről nyugat felé történt, és az intenzív keveredés ellenére sem jutott el Európába az összes kromoszómális DNS változat (Warmuth 2011, 2012).
Történelmi lómaradványok DNS vizsgálata:
Az elhalt maradványokból módszertani okokból többnyire csak az anyai ágú mtDNS szekvenciák vizsgálhatók. Cieslak és munkatársai 2010-ben 207 lómaradvány genetikai vizsgálatát végezték el. A maradványok egész Eurázsiából, Alaszkától az Ibériai félszigetig terjedő területekről származtak, és koruk a késői Pleisztocéntől (12 ezer éve) a Középkor végéig terjedt. Meglepő módon a legrégebbi maradványokban kontinens szerte ugyanazon anyai vonalakat találtá. Eszerint a 12 ezer éve élt vadló állomány egész Eurázsiában elterjedt, és már ebben az időben az ember beavatkozása nélkül is távoli területek állományai keveredtek. A Holocén kezdetén viszont a vadlovak Európában megritkultak, és elszigetelődtek egymástól az ázsiai az ibériai félszigeti populációk. A háziasítást megelőző korokból 87 anyai vadló vonalat azonosítottak, melyek közül a bronzkori lovakban 29, a vaskori lovakban 47, a középkori lovakban 21 volt jelen. Ebből arra következtettek, hogy a vaskorban ázsiában újra nagyszámú vadlovat fogtak be, és napjaink lóállományában az összes háziasított kanca vonalnak körülbelül a fele-harmada maradhatott fenn, a többi kihalt. A szerzők megerősítik, hogy a történelem során az anyai vonalak elterjedése igen gyorsan változott az újonnan befogott kancákkal, valamint távolról importált egyedek keresztezésével. Emiatt egyetlen terület lovai sem különülhettek el egymástól számottevően.
Keyser-Tracqui és munkatársai 2005-ben az Altáj hegység kurgánjaiba eltemetett 13 szkíta ló mtDNS szekvenciáit elemezték. Azt találták, hogy ezek olyan ma élő lovak anyai vonalaival mutatnak rokonságot, melyek a legkülönfélébb fajtákhoz tartoznak. Néhány minta tökéletes egyezést mutatott a kínai Guanzhong és Tuva lovak, valamint anatóliai lovak egyedeiből származó szekvenciákkal. Egy egyed szekvenciája tökéletes egyezést mutatott egy skandináv viking lómaradvány szekvenciájával, amit egy ma élő izlandi póniban is megtaláltak. A rokon szekvenciák közül azonban meglepő módon hiányzott az akhal-teke, amit a helyszín és a kultúrkör alapján eredetileg legvalószínűbbnek véltek. A szerzők arra konkludáltak, hogy az mtDNS szekvenciák alapján az anyai vonalak földrajzi eredetét, rokonsági és leszármazási viszonyait lehetetlen felderíteni, így a szkíta lovak története homályban maradt.
2010-ben Raskó István laboratóriumából Priskin Katalin munkásságának köszönhetően 17 avarkori és 14 honfoglaláskori lómaradvány mtDNS adatait közölték. A történeti adatok alapján vélhető rokonság miatt 24 ma élő akhal-teke, és 70 hucul egyed mtDNS szekvenciáját is meghatározták. Nagyjából ugyanarra az eredményre jutottak, mint a fentebb idézett munkák: “bizonyos esetekben negatív Fst-értékeket tapasztaltunk a populációk között. Ilyenkor az azonos populációhoz tartozó pár tagjaiban egyenként nagyobb a variancia, mint a két populáció között. Ezt a modell nem képes feloldani, tehát ebben az esetben nem állapítható meg a reális távolság a két mintacsoport között…….. Az avar, a honfoglalás kori, az akhal teke és a hucul lovak csoportját elemezve a genetikai variancia 96,52%-a a populációkon belüli varianciára vezethető vissza, a genetikai differenciálódás a csoportok között igen alacsony. Mivel a háziló fajtákon belüli variabilitása nagyon magas, nem különülnek el élesen az egyes fajták…… Arra következtethetünk, hogy önmagában a mitokondriális kontroll régió vizsgálata nem elegendő, és további markerekre is szükség van a lovak esetében a fajták elkülönítéséhez.”
Mivel a lovak csoportjait nem tudták elkülöníteni a továbbiakban azt keresték, hogy az egyedi szekvenciák az adatbázisok mely egyedi szekvenciáival (haplotípus) mutatnak legnagyobb egyezést. Azt találták, hogy 4 honfoglaláskori valamint 4 avarkori ló legközelebbi rokon szekvenciái (nem megegyező!!) akhal-teke egyedekben találhatók, valamint 7 avarkori ló azonos anyai vonalba tartozott 7 ma élő hucul egyeddel. A honfoglaláskori lovakban 5 olyan anyai is vonalat találtak, amelyek teljesen egyediek, egyetlen más lóból sem ismertek.
Ezeket az eredményeket semmiképp sem lehet úgy interpretálni, hogy a honfoglalók lovainak akár csak egy része akhal-teke lett volna, mivel mint azt a szerzők is, és a korábban bemutatott irodalmi hivatkozások is megerősítik, az mtDNS szekvenciákból nem következtethetünk fajtára. A négy akhal-tekével rokon szekvenciát mutató egyedről csak annyit mondhatunk, hogy valószínűleg egy több ezer éve élt közös ősanya leszármazottai. A 7 avarkori lóról azonban elmonható lett volna, hogy a ma élő huculok ősei lehettek, hiszen a szekvenciák megegyeztek! Az egyedeken látható tulajdonságok természetesen nem az mtDNS szekvenciáktól függenek, hanem a kromoszómális génektől melyek csak teljes genom szekvenálással hasonlíthatók össze. Ezért a honfoglaláskori lovakról többet elárulnak a csontmaradványok és a korabeli leírások, mint az mtDNS szekvenciák. A történeti adatok alapján valószínűsíthető a keleti “turáni” lófajtáktól való leszármazás, – mely az akhal-tekének is őse lehetett – de sokat mondó adat, hogy az avar és honfoglaláskori a lovaknak a legnagyobb példányai sen haladták meg a 150 cm-es marmagasságot, és többségük ennél jóval kisebb volt (Bartosiewicz 2009).
Teljes ló genom szekvenciák:
A teljes DNS állomány (genom) megszekvenálása még napjainkban is igen költséges. Elsőként 2009-ben közölték egy angol telivér kanca teljes genom szekvenciáját. Ezt követően 2012-ben hét másik ló részleges genom szekvenciája vált ismertté a következő fajtákból: andalúziai, arab, akhal-teke, izlandi póni, amerikai ügetőló (Standardbred), angol telivér, quarter horse. Ezeket a szekvenciákat számítógéppel összehasonlítva 50 ezer olyan pontot (nukleotidot) találtak, amelyek a vizsgált egyedek között változékonynak mutatkoztak. Pénzügyi okokból a további egyedekből már csak ezt az 50 ezer változékony pontot vizsgálták. Magyarán a további egyedekből már a genomoknak csak azt az egymilliomod részét hasonlították össze, amely várható különbségeket mutat az egyedek között. Ezt a vizsgálatot már 36 fajtához tartozó 814 lóval végezték el (Petersen 2013).
A teljes genomra kiterjedő összehasonlítással már DNS szinten is elkülöníthetővé váltak a fajták. Azonban megjegyzendő, hogy 7 egyed teljesen kilógott a képből vagyis azokat puszta DNS vizsgálat alapján más fajtához tartozónak vélnénk. Az is kiderült, hogy a lófajták DNS állománya sokkal kevésbé különbözik egymástól mint a többi háziállatok, például a kutyák fajtái. Legjobban a kevés egyedből származó vonalak (pl. Exmoor póni), a zárt törzskönyvű vonalak (pl. angol telivér), és az erősen szelektált vonalak (pl. ügetőló) mutatkoztak egységesnek, míg a nagy egyedszámú fajták (quarter horse) és a nagy ménesekben szabadon tartott ázsiai vonalak (pl. mongol, Tuva) kevésbé egységes képet mutattak.
A szekvenciák alapján felállítottak egy rokonságot tükröző törzsfát (lásd ábra). Ebben az egymáshoz hasonló DNS-ű lovak közel vannak egymáshoz.
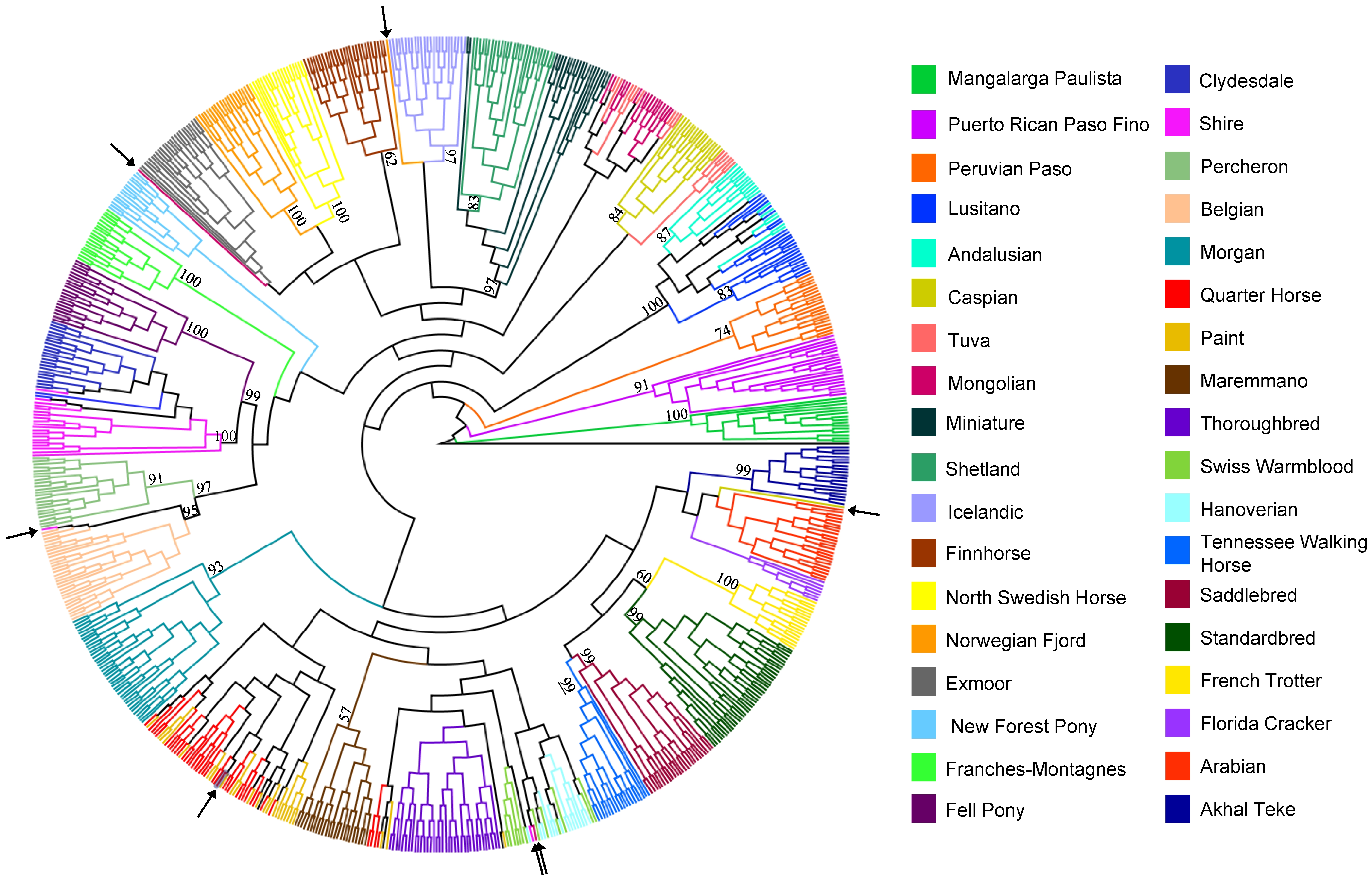
Az egy fajtához tartozó egyedek adatait matematikailag összevonva a vizsgált fajták az alábbi ábrán látható rokonsági viszonyokat mutatják: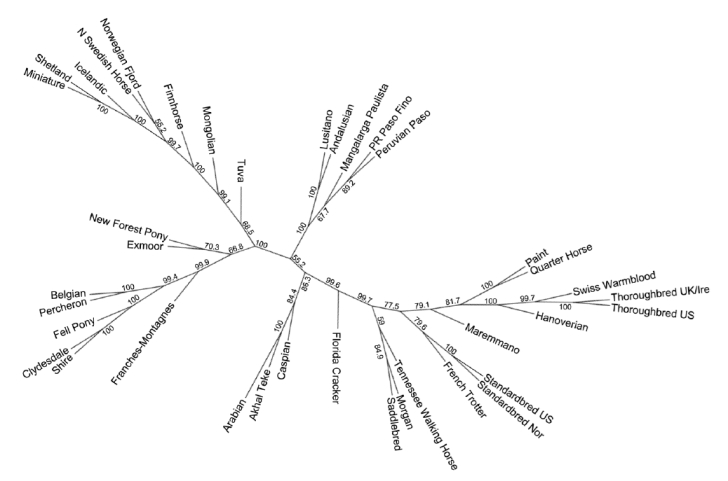
Összefoglalásként elmondható, hogy az egyedek és fajták tényleges rokonsági viszonyai csak az igen költséges teljes genom vizsgálattal válaszolhatók meg. Apai ágon szinte mindegyik ló egyetlen háziasított mén leszármazottja, anyai ágon pedig minden lófajta egyedei nagyszámú háziasított kancától származnak, és a fajták közötti különbséget jobbára csak az utóbbi évszázadok intenzív tenyésztői munkáinak köszönhetjük.
Idézett irodalom:
Achilli A., Olivieri A., Soares P. et al. (2012) Mitochondrial genomes from modern horses reveal the major haplogroups that underwent domestication. PNAS 109, 2449–54.
Bartosiewicz L: (2009) GONDOLATOK A „LOVAS NOMÁD” HAGYOMÁNYRÓL
http://www.shca.ed.ac.uk/staff/supporting_files/lbartosi/2009_Bartosiewicz_Nomadok.pdf
Bigi D1, Perrotta G, Zambonelli P. (2014) Genetic analysis of seven Italian horse breeds based on mitochondrial DNA D-loop variation. Anim Genet. 2014 Aug;45(4):593-5
Brandariz-Fontes C., et al (2013) Y-Chromosome Analysis in Retuertas Horses PLoS ONE 8(5): e64985.
doi:10.1371/journal.pone.0064985
Cieslak M., et al., (2010) Origin and History of Mitochondrial DNA Lineages in Domestic Horses PLoS
ONE 5(12): e15311. doi:10.1371/journal.pone.0015311
Han H., et al. (2015) Y-Single Nucleotide Polymorphisms Diversity in Chinese indigenous horses Asian Australas J. Anim. Sci. Anim. Sci.. http://dx.doi.org/10.5713/ajas.14.0784
Jansen T., et al. (2002) Mitochondrial DNA and the origins of the domestic horse PNAS vol. 99 10905–10910 www.pnas.org_cgi_doi_10.1073_pnas.152330099
Khanshour and Cothran (2013) Maternal phylogenetic relationships and genetic variation among Arabian horse populations using whole mitochondrial DNA D-loop sequencing BMC Genetics 2013, 14:83
http://www.biomedcentral.com/1471-2156/14/83
Lippold S., et al. (2011) Whole mitochondrial genome sequencing of domestic horses reveals incorporation of extensive wild horse diversity during domestication BMC Evolutionary Biology 2011, 11:328 doi:10.1186/1471-2148-11-328
Petersen et al. (2013) Genetic Diversity in the Modern Horse Illustrated from Genome-Wide SNP Data
PLoS ONE 8(1): e54997. doi:10.1371/journal.pone.0054997
Wallner B, et al. (2013) Identification of Genetic Variation on the Horse Y Chromosome and the Tracing of Male Founder Lineages in Modern Breeds. PLoS ONE 8(4): e60015. doi:10.1371/journal.pone.0060015
Warmuth V, et al. (2011) European Domestic Horses Originated in Two Holocene Refugia. PLoS ONE 6(3): e18194. doi:10.1371/journal.pone.0018194
Warmuth V. etal., (2012) Reconstructing the origin and spread of horse domestication in the Eurasian steppe.
Proc Natl Acad Sci U S A. 2012 May 22;109(21):8202-6. doi: 10.1073/pnas.1111122109. Epub 2012 May 7.